الیگودندروگلیوما
خلاصه مقاله
الیگودندروگلیوما، جزو تومورهای اولیه نورواپیتلیال سیستم عصبی مرکزی است.منشاء الیگودندروگلیوما، الیگودندروسیت ها، نوعی از سلول های گلیال مغز است. براساس کرایتریای WHO برای طبقه بندی تومورهای CNS، به 2 نوع، الیگودندروگلیومای درجه 2 و الیگودندروگلیومای درجه 3(اناپلاستیک) تقسیم میشوند.نسبت بروز آن در مردان به زنان، از 1.1 تا 2 متغییر است. به لحاظ هیستوپاتولوژیک در الیگودندروگلیوما، صفحه هایی از هسته های گرد ایزومورفیک به همراه سیتوپلاسم شفاف دیده میشود که با نام کلاسیک"تخم مرغ سرخ شده شناخته میشود. اجسام پساوما و کلسیفیکاسیون های منتشر در تومور وجود دارند.در الیگودنروگلیومای درجه 3، آتیپی هسته ای و نکروز، پرولیفراسیون میکروواسکولار، دانسیته ی بالای سلولی، اشکالی میتوز سلولی نیز دیده میشود.سی تی اسکن بدون کنتراست(NECT) برای ارزیابی علائم غیر اختصاصی سیستم عصبی مرکزی است.الیگودندروگلیوما در CT scan بصورت توده ی حاشیه اMRI وسعت تومور و ماهیت ارتشاحی آن را نشان دهد. در توالی T2 ، بصورت Hypointense و در توالی T1 بصورت، Hyperintense دیده می شود. تقویت شدن تومور با کنتراست اغلب در MRI بصورت هموژن یا Patchy و Ring enhancement نشان دهنده ی تهاجمی بودن تومور و پروگنوز بد آن است.ی هایپودنس یا ایزودنس در کورتکس و ماده ی سفید ساب کورتیکال دیده میشود.تشخیص براساس علائم بالینی، تصویربرداری، ارزیابی هیستوپاتولوژی و مارکر های ژنتیکی IDH1 و IDH2 است. تشخیص های افتراقی الیگوندروگلیوما شامل آستروسیتومای منتشر، گلیوبلاستوما، تومور های نورواپیتلیال دیس امبریوپلاستیک، گانگلیوما و تومورهای مولتی ندولار و واکوئل کننده ی نورونی است. علائم آن غیراختصاصی اند. شایع ترین علامت بالینی ان، تشنج پارشیال ساده یا کمپلکس یا جنرالیزه رخ میدهد.سردرد، تغیییر وضعیت ذهنی، سرگیجه، تهوع، شکایت بینایی و ضعف نورولوژیک فوکال از علائم دیگر است.بطور کلی هر فرد با تشنج جدید یا نقص عصبی فوکال باید تحت تصویربرداری CNS قرار گیرد. درمان الیگودندروگلیوما چندعاملی است و شامل روش های جراحی، شیمی درمانی و رادیوتراپی است. برداشت کامل تومور اولین انتخاب برای درمان اولیه ی الیگودندروگلیوما است و بیوپسی،برای افزایش Survival و تائید تشخیص تومور و نوع آن بکار می رود. تعیین نوع روش جراحی با توجه به محل تومور، بیماری های زمینه ای بستگی دارد. ادامه ی درمان بیمار براساس پروگنوز وی تعیین میشود: در صورت سن کمتر از 40 سال، الیگودندروگلیومای درجه 2 و یا عدم وجود اختلالات نورولوژیک در بیمار، ادامه ی درمان بصورت انتظاری یا رادیوتراپی و کموتراپی با تموزولوماید انجام میشود. ولی در صورت سن بالای 40 سال، الیگودندروگلیومای درجه 3، وجود اختلالات نورولوژیک و یا وجود باقی مانده ی تومور در CNS، ادامه ی درمان رادیوتراپی و کموتراپی با تموزولوماید است.
الیگودندروگلیوما(Oligodendroglioma)
مقدمه:
الیگودندروگلیوما، جزو تومورهای اولیه نورواپیتلیال سیستم عصبی مرکزی است. این تومور نادر بوده و حدوداً 5 درصد از تومور های اینتراکرانیال را در برمی گیرد. منشاء الیگودندروگلیوما، الیگودندروسیت ها، نوعی از سلول های گلیال مغز است. 90 درصد در ماده ی سفید سوپراتنتوریال معمولاً در لوب های فرونتال و تمپورال و 10 درصد در Posterior fossa و نخاع قرار دارند. همچنین رشد آهسته و ارتشاحی داشته و تمایل به درگیری لپتومننژ ها دارند. براساس کرایتریای WHO برای طبقه بندی تومورهای CNS، به 2 نوع، الیگودندروگلیومای درجه 2 و الیگودندروگلیومای درجه 3(اناپلاستیک) تقسیم میشوند.
اپیدمیولوژی:
میزان بروز آن، 0.2 در هر 100000 نفر است و سومین تومور شایع اینتراکرانیال پس از گلیوبلاستوما و آستروسیتومای منتشر است. اغلب در مردان دیده میشود و نسبت بروز آن در مردان به زنان، از 1.1 تا 2 متغییر است. 2 پیک سنی برای بروز این تومور وجود دارد: 1) دهه ی 4 تا 5 ام در بزرگسالان(شایعتر)، 2)از سنین 12 تا 16 سالگی در کودکان(کمترشایع).
الیگودندروگلیومای درجه 3 اغلب 10 سال دیرتر از الیگودندروگلیومای نوع 2 بوجود می آید.
اتیولوژی:
به لحاظ ژنتیکی، موتاسیون IDH1 یا IDH2 همراه با حذف بازوهای کروموزومی 1p و 19q در آن وجود دارد و از ترانسفوماسیون نئوپلاستیک الیگودندروسیت ها، سلول های گلیال مغزی بوجود می آید.
هیستوپاتولوژی:
به لحاظ هیستوپاتولوژیک در الیگودندروگلیوما، صفحه هایی از هسته های گرد ایزومورفیک به همراه سیتوپلاسم شفاف دیده میشود که با نام کلاسیک"تخم مرغ سرخ شده(Fried egg) شناخته میشود. شبکه ای از عروق خونی نازک در اطراف تومور وجود دارد که به عنوان الگوی سیم مرغی(chicken-wire) شناخته میشود. اجسام پساوما و کلسیفیکاسیون های منتشر در تومور وجود دارند.در الیگودنروگلیومای درجه 3، آتیپی هسته ای و نکروز، پرولیفراسیون میکروواسکولار، دانسیته ی بالای سلولی، اشکالی ا میتوز سلولی نیز دیده میشود.
علائم بالینی:
علائم آن غیراختصاصی اند. شایع ترین علامت بالینی ان، تشنج است که بصورت پارشیال ساده یا کمپلکس یا جنرالیزه رخ میدهد. و علائم دیگر شامل سردرد، تغیییر وضعیت ذهنی، سرگیجه، تهوع، شکایت بینایی و ضعف نورولوژیک فوکال است. طول مدت علائم قبل از تسخیص بسیار متغییر است و گاهی ممکن است پس از یک تشنج جدید، سردرد یا خونریزی و یا حتی پس از گذشت یک دهه از صرع تشخیص داده شود. بطور کلی هر فرد با تشنج جدید یا نقص عصبی فوکال باید تحت تصویربرداری CNS قرار گیرد.
تصویربرداری:
سی تی اسکن بدون کنتراست(NECT) اولین ابزار تصویر برداری برای ارزیابی علائم غیر اختصاصی سیستم عصبی مرکزی است.
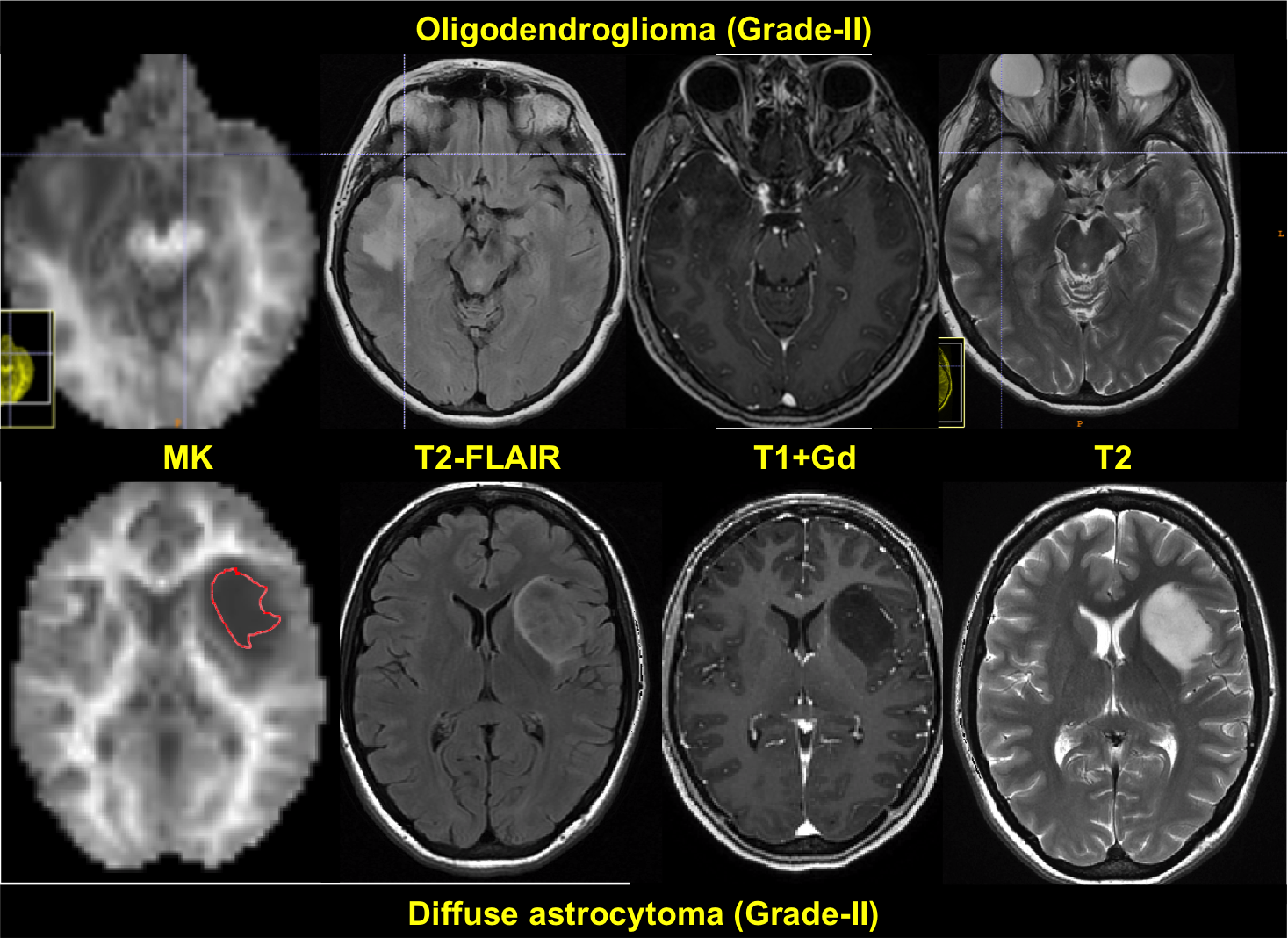
الیگودندروگلیوما در CT scan بصورت توده ی حاشیه ای هایپودنس یا ایزودنس در کورتکس و ماده ی سفید ساب کورتیکال دیده میشود. اغلب کلسیفیکاسیون نیز مشاهده میشود. تغییرات کیستیک، ادم اطراف تومور و خونریزی و Calvarial Remodelling نیز ممکن است مشاهده شود.
MRI بهتر می تواند وسعت تومور و ماهیت ارتشاحی آنرا نشان دهد. در توالی T2 ، بصورت Hypointense و در توالی T1 بصورت، Hyperintense دیده می شود.
تقویت شدن تومور با کنتراست اغلب در MRI بصورت هموژن یا Patchy و Ring enhancement نشان دهنده ی تهاجمی بودن تومور و پروگنوز بد آن است.
تشخیص:
تشخیص براساس علائم بالینی، تصویربرداری، ارزیابی هیستوپاتولوژی و مارکر های ژنتیکی IDH1 و IDH2 است.
تشخیص های افتراقی الیگوندروگلیوما شامل آستروسیتومای منتشر(Diffuse Astrocytoma)، گلیوبلاستوما(Glioblastoma)، تومور های نورواپیتلیال دیس امبریوپلاستیک، گانگلیوما و تومورهای مولتی ندولار و واکوئل کننده ی نورونی است.
مدیریت/درمان:
درمان الیگودندروگلیوما چندعاملی است و شامل روش های جراحی، شیمی درمانی و رادیوتراپی است.
برداشت کامل تومور اولین انتخاب برای درمان اولیه ی الیگودندروگلیوما است و برای اخذ بیوپسی، افزایش Survival و تائید تشخیص تومور و نوع آن بکار می رود. محل تومور، بیماری های زمینه ای و دیگر ریسک فاکتور های جراحی جهت تعیین نوع روش جراحی بکار می روند.
ادامه ی درمان بیمار براساس پروگنوز وی تعیین میشود: در صورت سن کمتر از 40 سال، الیگودندروگلیومای درجه 2 و یا عدم وجود اختلالات نورولوژیک در بیمار، ادامه ی درمان بصورت انتظاری یا رادیوتراپی و کموتراپی با تموزولوماید انجام میشود.
ولی در صورت سن بالای 40 سال، الیگودندروگلیومای درجه 3، وجود اختلالات نورولوژیک و یا وجود باقی مانده ی تومور در CNS، ادامه ی درمان رادیوتراپی و کموتراپی با تموزولوماید است.
منابع:
1)van den Bent, M. J., & Chang, S. M. (2018). Grade II and III Oligodendroglioma and Astrocytoma. Neurologic clinics, 36(3), 467–484. https://doi.org/10.1016/j.ncl.2018.04.005
2)Bou Zerdan, M., & Assi, H. I. (2021). Oligodendroglioma: A Review of Management and Pathways. Frontiers in molecular neuroscience, 14, 722396. https://doi.org/10.3389/fnmol.2021.722396
3)Weller, M., van den Bent, M., Preusser, M., Le Rhun, E., Tonn, J. C., Minniti, G., Bendszus, M., Balana, C., Chinot, O., Dirven, L., French, P., Hegi, M. E., Jakola, A. S., Platten, M., Roth, P., Rudà, R., Short, S., Smits, M., Taphoorn, M. J. B., von Deimling, A., … Wick, W. (2021). EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nature reviews. Clinical oncology, 18(3), 170–186. https://doi.org/10.1038/s41571-020-00447-z
4)Engelhard H. H. (2002). Current diagnosis and treatment of oligodendroglioma. Neurosurgical focus, 12(2), E2. https://doi.org/10.3171/foc.2002.12.2.3
5)Tork, C. A., & Atkinson, C. (2022). Oligodendroglioma. In StatPearls. StatPearls Publishing.
6)Wesseling, P., van den Bent, M., & Perry, A. (2015). Oligodendroglioma: pathology, molecular mechanisms and markers. Acta neuropathologica, 129(6), 809–827. https://doi.org/10.1007/s00401-015-1424-1
مقالات مرتبط
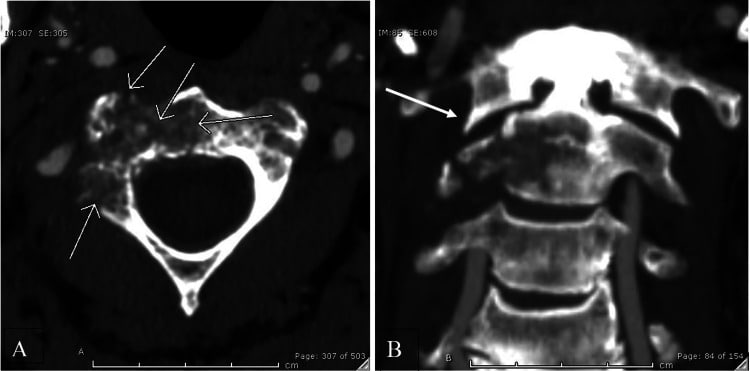
شکستگی C2
یک بیمار مرد 65 ساله به دلیل شروع حاد درد شدید در ناحیه کتف و گردن راست در بیمارستان بستری شد. درد علیرغم استفاده از داروهای بدون نسخه به مدت 10 روز پایدار بود. بیمار هیچ گونه بی حسی یا ضعف همراه را گزارش نکرد.
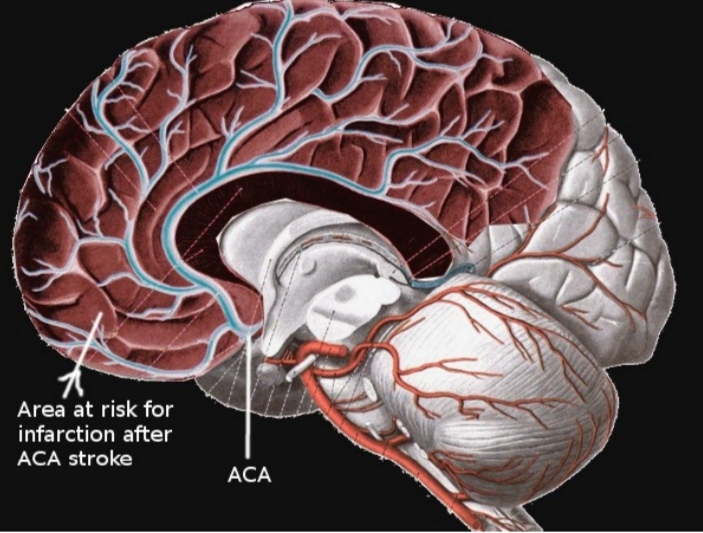
انفارکت ناشی از شریان مغزی قدامی (ACA)
انفارکت های شریان مغزی قدامی در قلمرو شریان مغزی قدامی رخ می دهد که شامل قسمت فوقانی و مدیال لوب تمپورال و نیز میدلاین لوب فرونتال می باشد. شریان ACA شاخه کاروتید داخلی بوده و خود به پنج سگمان با نام های A1 تا A5 تقسیم می شود. ساختار آناستوموزی قوی این شریان موجب می شود تا ریسک بروز انفارکت در این ناحیه پایین باشد